连接组学的新方法
伊利诺伊大学厄巴纳-香槟分校的研究人员开发出一种方法,旨在让神经科学中最困难的任务之一变得更快、更具可扩展性:绘制神经元彼此之间如何连接。该技术名为 Connectome-seq,利用 RNA 条形码为神经元标记,并识别细胞在突触处的连接位置。研究团队表示,这种方法以单突触分辨率捕捉了小鼠大脑中的数千条神经连接。
这项发表于 Nature Methods 的研究,将大脑绘图重新定义为一个测序挑战,而不是主要依赖成像的任务。这正是研究人员声称的核心进展。该方法不再主要依赖费时费力的切片、显微成像和手工重建,而是使用可通过测序流程读取的分子标签。
为什么绘制大脑如此困难
理解神经回路长期以来都受到大脑巨大复杂性的限制。传统绘图方法往往需要把脑组织切成极薄的切片,进行成像,然后再一块一块地重建。这个过程虽然有效,但速度慢、劳动强度高,而且很难扩展到大量细胞和突触。
更新的基于测序的方法通过一次标记许多神经元提高了通量,但据伊利诺伊团队称,这些工具通常只能显示神经元延伸到哪里,而不能明确它们在突触处究竟连接了哪些细胞。这一区别很重要。只有当研究人员不仅能识别邻近关系或投射模式,还能识别真实的细胞间通信连接时,线路图才真正有用。
伊利诺伊大学细胞与发育生物学教授、该研究的负责人 Boxuan Zhao 用工程学语言概括了这一挑战。他认为,如果研究人员不知道大脑是如何接线的,就无法充分理解其功能,优化模型,也无法修复疾病扰乱系统时出现的问题。

Connectome-seq 如何工作
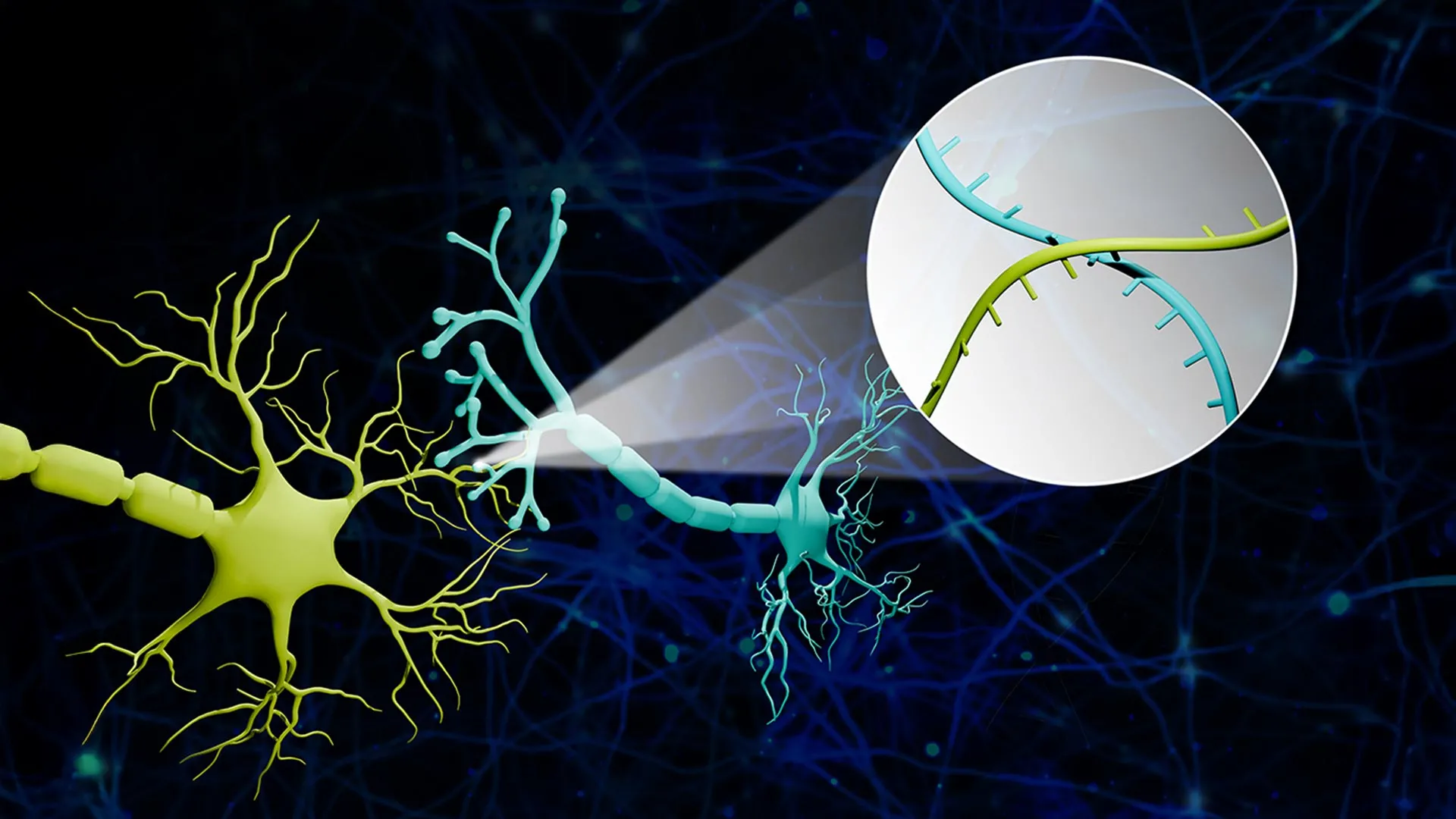
Connectome-seq 的基本概念是为每个神经元分配一个独特的分子条形码。随后,这些 RNA 条形码会在神经元彼此连接的地方相互混合,使研究人员能够通过读取成对的条形码信息来推断突触连接。最终得到的是一张由测序数据构建的连接图,而不是仅靠图像追踪得到的结果。
在学校提供的摘要中,研究人员将这项技术描述为能够以单突触分辨率同时绘制数千条神经连接。他们表示,这种速度、规模和细节的组合是现有技术无法提供的。
如果这一说法在更广泛应用中成立,那么这项方法可能标志着连接组学的重要一步,也就是构建越来越精确的神经网络地图的努力。这种意义不仅仅是技术层面的。更好的连接图可以揭示回路如何组织、信息如何在网络中流动,以及疾病可能如何改变正常结构。
小鼠实验显示了什么
在小鼠中,该方法据称发现了此前未知的大脑细胞连接。来源摘要并未列出这些具体连接,因此最稳妥、最有依据的结论是:研究团队发现了此前用现有方法未曾识别过的神经关系。
这很重要,因为任何绘图方法最强有力的检验之一,就是它是否揭示了会改变研究人员对系统理解的回路。只会重复已知结构的工具仍然有价值,但能够暴露新连接的工具,开始改变科学图景。
研究人员还强调,该方法具有单突触分辨率,这在该领域是一个重要表述。突触是神经元相互交流的功能接触点。在这个层面观察网络,能提高地图捕捉到有意义生物相互作用,而不只是宽泛解剖重叠的可能性。
为什么测序可能改变回路科学的速度
Connectome-seq 更深层的含义在于方法学。测序已经多次改变生物学,把曾经高度专门化的测量任务变得更便宜、更快、也更可扩展。伊利诺伊团队实际上认为,如果能够把连通性转化为条形码读出,那么回路绘图也可能经历类似转变。
这不会取消成像的重要性,后者在神经科学中仍然不可或缺,但它可以降低对传统连接组学流程中最慢部分的依赖。一种更适合测序的方式,可能让实验室研究更多细胞、比较更多条件,并在研究发育、学习、损伤或疾病时更快迭代。
它也可能让神经回路分析更好地融入现代分子生物学的其他部分,因为测序基础设施和数据管道已经在广泛建立。其战略价值不仅在于更高分辨率,还在于更高通量和更广泛的可及性。
疾病相关性是这一主张的重要部分
伊利诺伊研究人员明确将这项方法与神经系统疾病联系起来。Zhao 表示,该技术可直接用于理解神经退行性疾病中的回路功能障碍,并可为回路导向的治疗干预提供一个平台。
这是一个雄心勃勃的愿景,但方向很清楚。阿尔茨海默病和其他神经退行性疾病不仅是细胞死亡或蛋白质错误折叠的问题,也是回路失效的疾病。如果研究人员能够观察到特定连接如何丢失、重连或失稳,他们也许就能更好地识别早期变化,并设计更精准地针对受影响网络的干预措施。
摘要还暗示了其在早期检测中的潜在相关性,但并未宣称该方法本身就是临床诊断工具。在这一阶段,这项工作还是一种用于小鼠的研究平台,而不是人类医学应用。它当前的意义在于,作为基础和转化神经科学的赋能技术。
仍需证明什么
和许多技术突破一样,下一阶段是验证与推广。新的绘图方法必须证明可重复性、可控的错误率,以及在不同脑区和实验场景中的适用性。其他实验室的研究人员会想知道 Connectome-seq 可以多广泛地应用、实施起来有多难,以及它的读出与成熟方法相比如何。
来源材料支持这样一个强有力的说法,即该方法比传统方法更快、也更可扩展,但该领域最终会通过反复使用和比较来判断这一点。即便如此,这项工作仍符合生物学中的一个更大趋势:把结构转化为可以大规模读取、处理和分析的数据。
如果 Connectome-seq 能持续按描述运行,它或许能帮助大脑绘图从手工式重建,转向更高通量的回路科学。对神经科学而言,这将是一个有意义的转变。大脑的连线始终是理解其功能的核心。缺少的是一种能够大规模读取这些连线的实用方法。伊利诺伊团队的答案是给网络做标记,然后对代码进行测序。
本文基于 Science Daily 的报道。阅读原文.
Originally published on sciencedaily.com