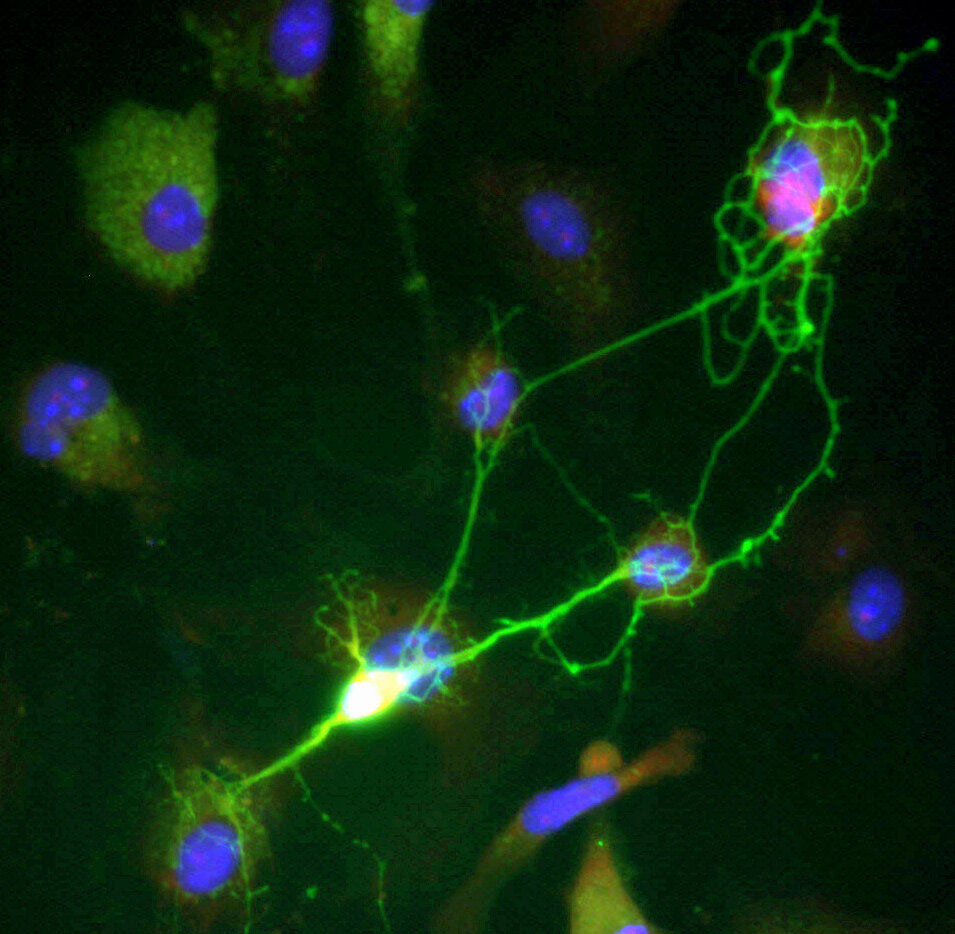
A targeted attempt to close a treatment gap
Cystic fibrosis is one of the best known inherited diseases, but treatment has remained uneven across the patient population. A study coordinated by the University of Trento describes a gene therapy strategy aimed at people whose disease is caused by the 1717-1G >A mutation, a group the researchers say accounts for about 10% of patients and has lacked an effective treatment option.
The work, published in Science Translational Medicine, focuses on a defect that prevents production of the CFTR protein. That protein is essential for moving chloride and bicarbonate ions across the surface of the lung epithelium, where it helps regulate mucus hydration and clearance. When CFTR is missing or not working properly, the disease can damage multiple organs, with lung complications remaining the leading cause of mortality.
What the study says
According to the University of Trento team, the new approach uses messenger RNA to deliver a genome-editing strategy designed to permanently correct the disease-causing mutation. The researchers describe the method as a way to repair the single defective DNA letter responsible for this form of cystic fibrosis.
That detail matters because the study is not simply another report on symptom management. It is framed as an effort to directly fix the underlying mutation in patients who do not benefit from some of the pharmacological therapies already available for other forms of the disease.
The study was coordinated by Anna Cereseto with Alessandro Umbach as co-corresponding author. In the description provided, the researchers present the therapy as both effective and potentially durable, raising the prospect of a treatment that could move beyond chronic disease control.
Why this matters
Cystic fibrosis treatment has advanced significantly over the past decade, but the field has also been defined by a persistent split: some patients can use highly effective mutation-specific drugs, while others remain outside that therapeutic progress. The mutation highlighted in this study falls into that second group.
That makes the Trento result notable even before any future clinical milestones are considered. It points to a route for precision treatment where the genetic cause is not merely identified but edited. If such an approach proves safe and effective beyond the current study, it could change how researchers think about other rare or poorly served cystic fibrosis variants as well.
The work also reflects a broader shift in medicine. Messenger RNA has already become an important delivery platform in other areas of biotechnology. Here, it is being positioned as part of a genome-editing toolkit, suggesting another pathway for mRNA beyond vaccines and transient protein expression.
From disease management to genetic repair
The distinction between managing cystic fibrosis and correcting it is central to the significance of this report. Existing therapies can be transformative, but they generally depend on the specific mutation involved and often require ongoing treatment. The Trento team describes its method as a permanent correction, which is a much more ambitious clinical claim.
At the same time, the study is best understood as a research milestone rather than a finished therapy. The source text points to promising results and a potential turning point, but it does not claim the treatment is already available to patients. What it does establish is that a credible academic group has published a peer-reviewed attempt to repair a mutation that has remained difficult to address.
For families affected by cystic fibrosis, that is the practical importance of the study. It expands the conversation from “which patients respond to current drugs” to “which mutations can be edited directly.”
What comes next
The most important next step will be whether this strategy can continue through the translational path that turns a laboratory result into a real therapy. Questions of delivery, durability, and safety will determine whether the approach can move toward human use. None of those issues are trivial for genome editing.
Still, the study adds meaningful momentum to a part of cystic fibrosis research that needs it most: therapies for patients left behind by existing mutation-specific medicines. Even if further work is required, the result strengthens the case that this treatment gap is not permanent.
In that sense, the paper stands out less as a generic biotech advance than as a focused attempt to bring precision medicine to a population that has had fewer options. For a disease studied as intensively as cystic fibrosis, reaching the remaining untreated mutations is the next hard frontier. This study suggests that frontier may be starting to move.
This article is based on reporting by Medical Xpress. Read the original article.
Originally published on medicalxpress.com