Un nouveau génotype H5N1 a progressé rapidement le long des couloirs migratoires
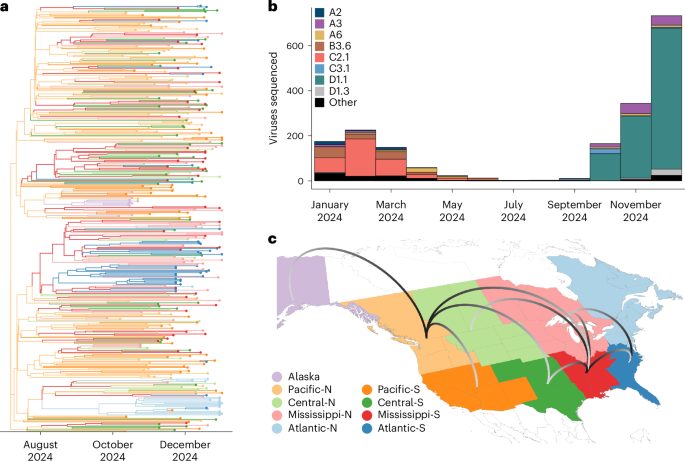
Une étude publiée en ligne dans Nature Medicine le 15 avril 2026 rapporte qu’un génotype nouvellement classé de grippe aviaire hautement pathogène, D1.1, s’est rapidement développé chez les oiseaux sauvages en Amérique du Nord pendant la saison migratoire de 2024. L’article décrit le virus comme un réassortant, détecté pour la première fois en septembre 2024, puis suivi grâce à des programmes de surveillance active et passive au Canada et aux États-Unis.
Le constat central n’est pas seulement que le H5N1 est resté présent dans les populations d’oiseaux sauvages, mais qu’un génotype distinct semble s’être propagé assez vite pour remplacer des lignées A(H5) antérieures dans plusieurs couloirs migratoires. C’est important, car les couloirs migratoires sont les voies par lesquelles la grippe aviaire peut se déplacer sur de longues distances, franchir des juridictions et réensemencer à plusieurs reprises des foyers dans de nouveaux endroits.
En reliant la surveillance génomique au mouvement saisonnier des oiseaux sauvages, l’étude offre une image plus précise de la manière dont une lignée virale spécifique peut passer de son émergence à une large extension géographique en peu de temps. Elle montre aussi combien il est crucial de maintenir des systèmes de surveillance capables de détecter ces changements avant qu’ils ne deviennent visibles dans le cheptel ou dans les comptages de cas humains.
Ce que les chercheurs ont rapporté
Selon le résumé fourni avec le candidat, des virus de grippe aviaire hautement pathogène A(H5N1) du clade 2.3.4.4b sont arrivés en Amérique du Nord fin 2021, puis se sont rapidement réassortis avec des virus aviaires locaux. Le nouveau génotype D1.1 a été détecté en septembre 2024. À l’aide de données de surveillance provenant du Canada et des États-Unis, les chercheurs ont suivi son apparition et sa diffusion pendant la migration automnale.
L’étude indique que l’analyse phylodynamique a montré que les virus D1.1 formaient un groupe monophylétique. En pratique, cela soutient l’idée que les virus suivis dans le réseau de surveillance appartenaient à une lignée cohérente, récemment développée, plutôt qu’à un ensemble dispersé de détections non liées. L’article précise en outre que D1.1 a remplacé des génotypes A(H5) antérieurs dans plusieurs couloirs migratoires, soulignant qu’il ne s’agissait pas d’un événement marginal aux confins de la carte de surveillance.
Le texte source relie aussi l’expansion de D1.1 à des détections chez d’autres hôtes, dont 17 cas humains, quatre sévères ou mortels. Dans le même temps, le résumé note que les marqueurs d’adaptation aux mammifères observés dans les cas humains n’ont pas été détectés dans les virus d’oiseaux sauvages analysés dans l’étude. Cette distinction est importante : elle suggère que les résultats de surveillance chez les oiseaux sauvages n’ont pas montré directement les mêmes signatures d’adaptation que celles rapportées dans les cas humains.

Pourquoi cela dépasse la seule surveillance aviaire
L’étude se situe à l’intersection de l’écologie de la faune sauvage, de la santé animale et de la santé humaine. Un génotype en expansion rapide chez les oiseaux sauvages n’est pas seulement une question de conservation ou de vétérinaire ; c’est aussi un avertissement sur la vitesse à laquelle l’écologie de la grippe peut évoluer. Lorsqu’une lignée s’installe le long des couloirs migratoires, le nombre d’occasions de passages à d’autres espèces, d’exposition agricole et de transmission interespèces augmente.
Le résumé n’affirme pas que D1.1 ait lui-même acquis les marqueurs d’adaptation aux mammifères observés dans les infections humaines, et cette prudence fait partie du message. Le risque grippal est façonné par une combinaison mouvante de génétique virale, d’exposition de l’hôte et d’opportunité écologique. Un génotype peut devenir épidémiologiquement important sans montrer immédiatement toutes les mutations associées à l’adaptation chez l’être humain.
Cela rend la surveillance génomique précoce et étendue particulièrement précieuse. Le fait que l’article combine surveillance active et passive montre qu’aucune méthode d’échantillonnage unique ne suffit pour un virus capable de circuler à l’échelle continentale. Détecter un nouveau génotype n’est qu’une étape. Comprendre s’il remplace d’autres lignées, à quelle vitesse il se déplace et s’il apparaît chez d’autres hôtes nécessite un échantillonnage soutenu.
Ce que l’article dit et ne dit pas
Le matériau source permet plusieurs conclusions claires. D1.1 est apparu comme un réassortant nouvellement détecté en septembre 2024. Il s’est rapidement propagé chez les oiseaux sauvages pendant la migration automnale de 2024. Il a formé un groupe monophylétique dans l’analyse des auteurs et a remplacé des génotypes A(H5) antérieurs dans plusieurs couloirs migratoires. Le résumé ajoute que les virus candidats vaccinaux ont conservé une réactivité croisée antigénique avec les souches D1.1.
Ce dernier point est notable, car il suggère que, d’après les résultats rapportés par les auteurs, les candidats vaccinaux n’ont pas perdu leur pertinence antigénique avec l’ascension de ce génotype. Cela n’élimine pas le risque, mais indique que l’évolution virale et la préparation vaccinale n’ont pas immédiatement divergé de la manière que redoutent le plus les autorités sanitaires.
Le résumé ne fournit pas de répartition géographique complète, de chronologie mois par mois du remplacement ni de détails complets sur le contexte des cas humains. Ces éléments peuvent figurer dans l’article complet, mais ils ne sont pas inclus dans le texte source fourni. Ce que l’on peut affirmer avec certitude, c’est que l’étude documente une reconfiguration rapide et marquante du paysage H5N1 chez les oiseaux sauvages d’Amérique du Nord au cours d’une seule saison migratoire.

Un signal pour l’ère de la surveillance
La leçon plus large est que la surveillance de la grippe ressemble de plus en plus à une course contre la recombinaison et le déplacement viral. Au moment où une lignée est largement discutée dans l’espace public, elle a peut-être déjà traversé plusieurs couloirs migratoires et touché plusieurs hôtes. Le rapport sur D1.1 montre pourquoi le suivi génomique est devenu une infrastructure essentielle, et non un simple outil de recherche de niche.
Pour les décideurs et les agences de santé, l’étude renforce un message familier mais toujours urgent : les menaces émergentes de grippe apparaissent souvent d’abord dans les systèmes écologiques, et non dans les hôpitaux. Pour les chercheurs, elle offre une étude de cas sur la rapidité avec laquelle une lignée réassortante peut s’établir. Et pour le grand public, elle rappelle que les récits sur la grippe aviaire ne sont plus de simples incidents isolés de fermes ou de faune sauvage. Ce sont des événements de systèmes continentaux qui exigent une attention soutenue bien avant qu’un quelconque bilan de cas humains n’apparaisse en une.
Cet article est basé sur un reportage de Nature Medicine. Lire l’article original.
Originally published on nature.com


